Article Text

Abstract
Background SARS-CoV-2 can spread rapidly on maritime platforms. Several outbreaks of SARS-CoV-2 have been reported on warships at sea, where transmission is facilitated by living and working in close quarters. Core components of infection control measures such as social distancing, patient isolation and quarantine of exposed persons are extremely difficult to implement. Whole genome sequencing (WGS) of SARS-CoV-2 has facilitated epidemiological investigations of outbreaks, impacting on outbreak management in real time by identifying transmission patterns, clusters of infection and guiding control measures. We suggest such a capability could mitigate against the impact of SARS-CoV-2 in maritime settings.
Methods We set out to establish SARS-CoV-2 WGS using miniaturised nanopore sequencing technology aboard the Royal Fleet Auxiliary ARGUS while at sea. Objectives included designing a simplified protocol requiring minimal reagents and processing steps, the use of miniaturised equipment compatible for use in limited space, and a streamlined and standalone data analysis capability to allow rapid in situ data acquisition and interpretation.
Results Eleven clinical samples with blinded SARS-CoV-2 status were tested at sea. Following viral RNA extraction and ARTIC sequencing library preparation, reverse transcription and ARTIC PCR-tiling were performed. Samples were subsequently barcoded and sequenced using the Oxford Nanopore MinION Mk1B. An offline version of the MinKNOW software was used followed by CLC Genomics Workbench for downstream analysis for variant identification and phylogenetic tree construction. All samples were correctly classified, and relatedness identified.
Conclusions It is feasible to establish a small footprint sequencing capability to conduct SARS-CoV-2 WGS in a military maritime environment at sea with limited access to reach-back support. This proof-of-concept study has highlighted the potential of deploying such technology in the future to military environments, both maritime and land-based, to provide meaningful clinical data to aid outbreak investigations.
- COVID-19
- molecular diagnostics
- molecular biology
Data availability statement
Data are available upon reasonable request. Data are available on reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
It is known that SARS-CoV-2 causes outbreaks on military maritime platforms which can be difficult to control.
Whole genome sequencing (WGS) is a valuable tool which can impact outbreak management by identifying transmission patterns, infection clusters and guiding control measures.
WHAT THIS STUDY ADDS
This proof-of-concept study describes establishing SARS-CoV-2 WGS using miniaturised nanopore sequencing technology with standalone data processing aboard a ship at sea for the first time.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
This approach has the potential to be used in multiple deployed military settings, not only for SARS-CoV-2, but also in targeting other pathogens, providing a valuable additional diagnostic capability.
Introduction
The pandemic caused by SARS-CoV-2 has resulted in several challenges for military populations, with outbreaks reported in numerous settings ranging from training establishments to operational deployments.1–3 Within these settings, environmental constraints may facilitate viral transmission and impair effective infection prevention and control measures. This has been especially true for maritime platforms, which is consistent with previous experience of respiratory viruses (eg, influenza) and enteric pathogens (eg, norovirus) which can spread rapidly.4 5 Military populations at sea may be sizeable, with many thousands of sailors deployed at any one time, presenting further challenges.6 In addition, naval commands have had to balance operational capability with the management of a highly contagious and morbid respiratory virus while individuals work in proximity and in confined spaces.7
Several outbreaks of SARS-CoV-2 have occurred on warships at sea, where the core components of infection control measures, such as social distancing, isolation of sick patients and quarantine of exposed persons, are extremely difficult to implement.6 8–10 SARS-CoV-2 can spread quickly among a ship’s crew with transmission readily facilitated by close-quarters conditions and by asymptomatic and pre-symptomatic infected crew members.6 The index case(s) of SARS-CoV-2 aboard a ship may originate from initial embarkation or following acquisition by crew members during overseas port visits.9 The latter raises the potential for individuals being exposed to novel viral variants, including vaccine escape mutants, posing a greater risk to personnel and the subsequent mission.
In March 2020, the UK government launched the COVID-19 Genomics UK Consortium (COG-UK) comprising of the NHS, public health agencies and academic partners, with the objective of sequencing the genomes of SARS-CoV-2.11 Whole genome sequencing (WGS) provides high-quality consensus sequences of SARS-CoV-2 allowing high-resolution analysis.12 This includes not only monitoring viral evolution and the identification of new variants, but also facilitating genomic epidemiological investigations of outbreaks through the characterisation of transmission dynamics.13 14
WGS has previously been used retrospectively to supplement traditional outbreak investigations. However, this is the first pandemic where WGS technology has been widely available, allowing for its use in a variety of scenarios including outbreaks in healthcare settings, long-term care facilities, educational and correctional establishments.12 15–17 When used in a clinically relevant time frame, particularly if contemporaneous, WGS can directly impact outbreak management in real time by elucidating transmission patterns, identifying clusters of infection and guiding the implementation of control measures to prevent further disease spread.18
We proposed that naval commands can be supported to mitigate against the impact of SARS-CoV-2 on the delivery of operational capability through the development of a WGS capability in the maritime setting. Therefore, we set out to establish SARS-CoV-2 WGS using miniaturised nanopore sequencing technology aboard the Royal Fleet Auxiliary (RFA) ARGUS while at sea. Our objectives included designing a simplified protocol requiring minimal reagents and processing steps, the use of miniaturised equipment compatible for use in the limited space aboard a ship, and a streamlined and standalone data analysis capability to allow rapid in situ data acquisition and interpretation. Here we describe the delivery of such a capability.
Materials and methods
Maritime setting
RFA ARGUS is the Royal Navy’s Maritime Role 3 (MR3) medical treatment facility. In its largest configuration, the MR3 can provide over 100 beds, including 4 resuscitation bays, 4 operating tables, 10 intensive care beds, 20 high dependency beds and 70 general ward beds.19 Clinical diagnostic support includes both radiology (portable X-ray, ultrasound and 64-slice CT) and comprehensive pathology services. The pathology capability, led by consultant clinicians and delivered by military biomedical scientists (BMS), consists of haematology, biochemistry, blood transfusion (including massive transfusion with an emergency donor panel) and microbiology services, which are provided from two dedicated laboratories.
In March 2022, following a maintenance period, RFA ARGUS sailed from Devonport naval base, Plymouth, to conduct Exercise MEDICAL RESPONSE, the aim of which was to train, assure and externally validate the MR3 workforce and processes to allow declaration of operating capability to Navy Command and the Ministry of Defence. This was the setting for establishing and trialling SARS-CoV-2 WGS in the microbiology laboratory aboard the MR3.
Clinical samples
Clinical samples (n=11) were stored residual upper respiratory tract specimens previously identified as positive for SARS-CoV-2 by PCR. The lineage and variant of SARS-CoV-2 had been determined. Each sample was anonymised (both patient origin and SARS-CoV-2 status) and assigned a unique identifier prior to use in this study.
Sequencing
For sequencing studies, the MinION Mk1B was used, coupled to a standalone deployable encrypted laptop with excess of minimum system requirements for analysis and without internet connectivity. The deployable sequencing module was developed in consultation with colleagues in the Royal Navy, UK Defence Medical Services Pathology, and the University of Birmingham. Equipment and consumable requirements were developed from the ARTIC Network for Ebola virus Field Sequencing.20 Extraction of viral RNA was performed using the QIAamp Viral RNA Extraction Kit in accordance with the manufacturer’s instructions (Qiagen, UK). The protocol for ARTIC sequencing library preparation was adapted from elements of the LoCost protocol using the NEBNext ARTIC SARS-CoV-2 Workflow for Oxford Nanopore Technologies Sequencing (New England BioLabs, UK).21 Reverse transcription and ARTIC PCR-tiling were performed using the NEBNext companion kit reagents and a MiniPCR (Cambio, UK). Samples were barcoded using the Native Barcoding Expansion 96 EXP-NBD196 (Oxford Nanopore Technologies, UK) and barcoded samples were sequenced in groups of a maximum of 12. During the development phase (initial verification of the workflow), specimens were sequenced in duplicate (two duplicate specimens per barcode) to demonstrate workflow functionality. During the subsequent deployed trial phase at sea, singlet sequencing of up to 12 samples in a pool, including a non-template control (NTC), was performed. For ligation of barcodes and adapters, the Ligation Sequencing Kit (SQK-LSK109) was used in accordance with the manufacturer’s instructions (Oxford Nanopore Technologies, UK). For data acquisition, the Flow Cell (R9.4.1) FLO-MIN106D was used and prepared with the Flow Cell Priming Kit EXP-FLP002 prior to flow cell loading (Oxford Nanopore Technologies, UK). The library was loaded in accordance with the manufacturer’s instructions. Due to limited internet connectivity and security precautions, an offline version of the MinKNOW software (V.21.10.4) was used (kindly provided by Oxford Nanopore Technologies, UK). The analysis run was set up with default settings tailored to the use of the kits described above but was modified to set basecalling mode to high-accuracy basecalling and setting the length of time for sequencing to 12 hours. Basecalled Fastq files were imported into the CLC Genomics Workbench for downstream analysis (Qiagen, UK). The CLC Genomics Workbench used the onboard Long Read Sequencing Plug-in with the supplier-provided ARTIC sequencing pipeline. Validation studies were performed using the Epi2Me (Oxford Nanopore Technologies, UK) online toolkit, to perform FastQC and the ARTIC workflow, including downstream analysis with NextClade and Pangolin for molecular epidemiological analysis of SARS-CoV-2 genomes.22 23 Further quality control was performed within the CLC Genomics Workbench, including coverage assessment and validation, using anonymised and trimmed data in the Stanford Coronavirus Resistance Database online tool.24 Local alignment was performed using the CLC Genomics Workbench using MUSCLE and phylogenetic tree construction by maximum likelihood (ML) analysis and the General Time Reversible (GTR) evolutionary model.
Results
Establishing a sequencing module for maritime deployment
As a proof-of-concept study designed to illustrate the use of this technology in a military maritime setting, equipment considerations included ease of use, robustness and size. Prior to deployment, the more sensitive equipment (eg, thermocyclers and MinION Mk1B) and an encrypted laptop were loaded into a secure transport case containing shock-absorbent modular padding (Pelicase, RS Components, UK). Frozen and refrigerated reagents were consolidated into appropriate temperature-controlled boxes for transportation to RFA ARGUS where they were removed and stored on arrival.
The laboratory space provided was minimal, consisting of a single bench space, airflow controls, a laboratory hand-wash basin and space for routine microbiology clinical testing (Figure 1A). Most of the bench space was used for post-PCR amplification sample handling, which presents a significant risk of contamination of surfaces that could interfere with subsequent testing. To reduce this risk, a pop-up hood was used, together with a timed ultraviolet lamp to allow decontamination of surfaces by removing residual DNA and RNA. Nuclease spray was also routinely used to further minimise the risk of contamination (Figure 1B,D). Small deployable thermocyclers were used to perform the PCR-tiling protocol as part of the ARTIC sequencing process. The ARTIC procedures, pipelines and recommendations have been in development for several years, with the most recent application to create a PCR-tiling solution for SARS-CoV-2 sequencing. As this was the approach taken by the large academic and public health consortia in the UK (COG-UK), the decision was made to adopt this same approach for sequencing in this study. The analytical equipment, including the laptop and MinION Mk1B, was set up separate from both the initial sample preparation area and the post-PCR handling hood (Figure 1C,E). Despite the environmental and technical challenges, the equipment was readily established in the maritime setting.
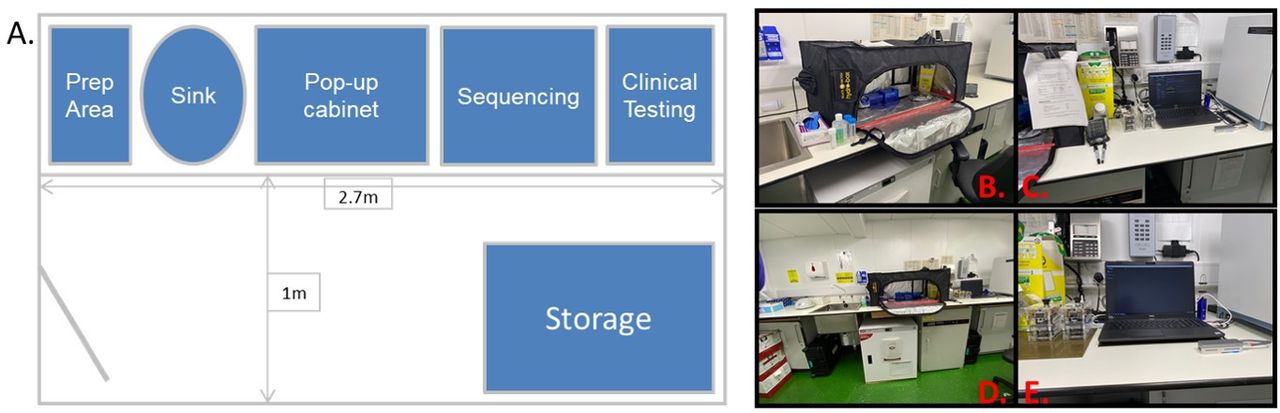
Laboratory design for SARS-CoV-2 WGS aboard RFA ARGUS. (A) Schematic diagram of the laboratory location, with approximate measurements of available space, illustrating the challenging setting for use to provide this complex workflow in a deployable and adaptable setting. (B) Pop-up hood used for sample processing to prevent contamination and enable easy decontamination to take place. (C) Sequencing and amplification equipment set-up for use. (D) Wide-angle image of the available bench space and limited storage. (E) MinION Mk1B and laptop. RFA, Royal Fleet Auxiliary; WGS, whole genome sequencing.
A detailed standard operating procedure was designed to enable the operator to conduct a streamlined workflow without readily available access to expert support (as described within the Methods section), with explanations given to aid understanding by unfamiliar operators. While UK military BMS are highly qualified to deliver a deployed pathology capability, they typically have limited experience of molecular biology and next-generation sequencing. Therefore, on completion of the initial development phase and prior to deployment (trial phase), an instructional briefing was delivered together with 2 days of practical training period to the BMS, consisting of a practice work-through of the protocol and a trial protocol on control specimens. Detailed instructions were provided to deploying BMS.
Development phase studies demonstrated a robust workflow suitable for the deployment
Initial work consisting of trialling the well-described ARTIC protocol using commercially available reagents from New England BioLabs, according to the manufacturer’s instructions, demonstrated results were reliably obtained and of the quality expected based on acceptability criteria defined by the ARTIC Network (Figure 2). Local verification of the CLC Genomics Workbench ARTIC sequencing pipeline was performed with Qiagen support. A selection of 15 previously analysed specimens, originally sequenced by Johns Hopkins University (JHU), USA, was provided for validation studies and to aid software familiarisation. The outcome of this initial verification of the software was acceptable based on 100% concordance between JHU variant classification and the analysis of the data from the CLC Genomics Workbench and the software was considered suitable for use in a deployed setting as it enabled the functionality required in the field without requiring remote support or online tools for analysis. To provide reassurance that the CLC Genomics pipeline produced genome sequences which were in line with outputs from more conventional analysis tools for SARS-CoV-2, the development phase samples, and later the trial phase sample data, were reanalysed using the Epi2Me platform provided by Oxford Nanopore Technologies, and analysed using the ARTIC pipeline provided. Further lineage and clade confirmation was achieved by reanalysis of consensus sequences from the CLC Genomics Workbench using the Stanford mutation analysis tool for SARS-CoV-2, which showed the clade/variant calling was 100% concordant in all cases.

Tissue culture-derived control samples were used for initial testing of the workflow comprising duplicate specimens of Alpha variant virus (B.1.1.7) and one replicate of Gamma variant (P.1.12); all three specimens were sequenced in parallel. Lineage was called using the Pangolin software and clade (variant) type was called using NextClade in the ARTIC workflow. The proportion of non-called bases is shown, indicated by an ‘N’. This is displayed where the number of reads covering a region of the genome (read depth) is <100. A total yield from the MinION Mk1B is shown in mega-bases (Mbases) to illustrate run yield.
Quality control and analysis of sequencing data
A proficiency panel of 12 specimens was created consisting of 11 residual samples collected between September 2021 and February 2022 at the University Hospitals Birmingham NHS Foundation Trust, Birmingham, UK and a single NTC. Only specimens with a Ct value of <30 (determined by testing on the Alinity m SARS-CoV-2 assay; Abbott Molecular, UK) were selected for inclusion, in accordance with guidance from COG-UK to minimise risk of sequencing failure. All specimens in the panel positive for SARS-CoV-2 had been previously sequenced by the UK Health Security Agency Laboratory, Birmingham, as part of routine surveillance. Based on recommendations from Oxford Nanopore Technologies and the development phase data obtained, 12 samples were selected as these could be run as a single pool on the flow cell, and would enable relatedness studies to be performed. The proficiency testing panel was randomised and blinded to the trial team aboard RFA ARGUS.
The proficiency panel consisted of nine samples collected from unrelated hospital-onset cases, typed as Delta variant, and two samples, typed as Omicron variant, which were expected to show high relatedness phylogenetically due to close epidemiological links. One negative control sample was provided as the NTC.
The interpretation of data obtained from the deployed sequencing matched that already known from routine typing/sequencing previously performed under COG-UK (Table 1). The data were reanalysed using Epi2Me through the ARTIC pipeline to calculate quality scores and validate the data produced in trials at sea. Nine of 11 specimens had good-quality scores according to the ARTIC analysis pipeline in Epi2Me, with sufficient coverage of ≥97%. Two samples (1 and 2) had reduced coverage of 65% and 72%, respectively, reducing quality scores, labelling them as poor quality. A lineage was not assigned but despite this, the Epi2Me software successfully managed to identify the correct variant based on analysis. These poor-quality samples were removed from further relatedness studies due to reduced coverage. This is despite these samples having a low Ct, and is most likely attributable to storage conditions and effect of freeze–thaw cycles compromising integrity of viral RNA. Notably, there was 100% concordance between variant typing performed during trials at sea, compared with the validation analysis performed on return, and in comparison with the initial variant typing data assigned when sequenced through COG-UK. The hands-on time of the military BMS to conduct this work, from starting the RNA extraction to loading MinION Mk1B, was approximately 8 hours followed by 12 hours for a sequencing run.
Details of the 12 samples established as a proficiency testing panel for this study showing sequencing data generated during the deployed phase at sea which was reanalysed using the Epi2Me toolkit and the ARTIC pipeline
Molecular epidemiological analysis performed at sea
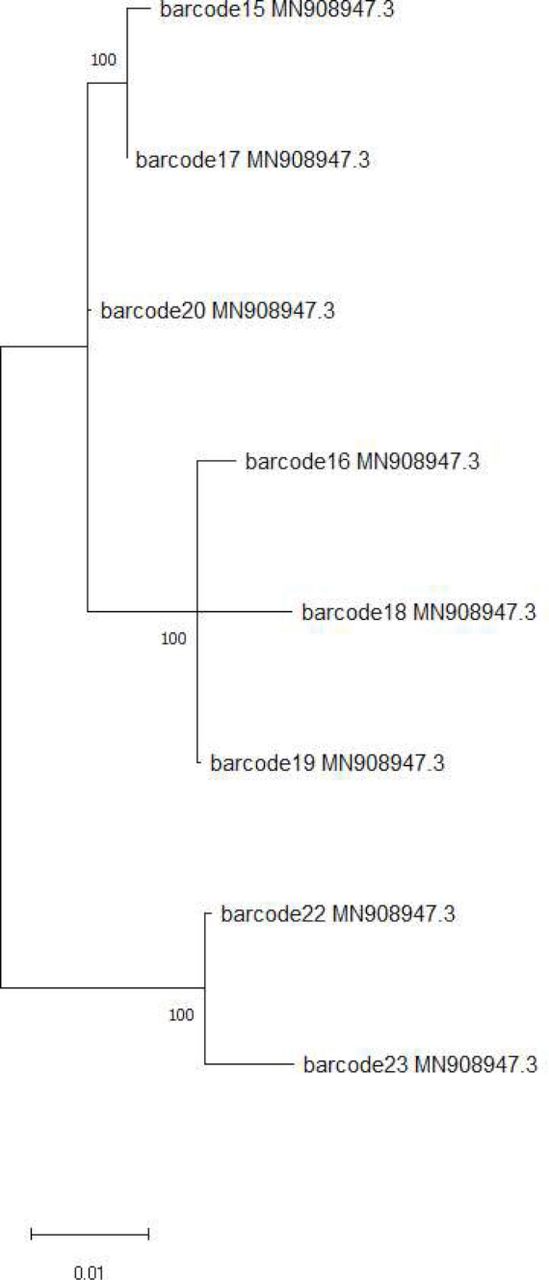
To evaluate the use of the CLC Genomics Workbench to enable local relatedness studies between available isolates, the data acquired from sequencing of the proficiency testing panel were subjected to ML analysis using the GTR model of evolution (Figure 3). ML analysis helps to describe the relatedness in genome sequences given in the tree, by finding the phylogenetic tree characteristics which best match the probability of the sequences being more or less closely related. As 11 samples had been sequenced in a pool of 12, this meant all 11 could be analysed together. For construction of the ML phylogenetic tree, samples with coverage of <98% were excluded (three specimens), resulting in analysis of the remaining sample data (eight specimens). This analysis demonstrated expected relatedness based on previously identified epidemiological associations, including eight unrelated cases from November to December 2021, which all sequenced and genotyped locally as the Delta variant of SARS-CoV-2, and two isolates from hospital-onset cases where a direct patient-to-patient transmission event was suspected epidemiologically in February 2022, and both had previously sequenced and genotyped as Omicron variants. Original phylogenetic or sequencing data were not available for review for comparative assessment against reanalysis during this study but overall, the relatedness demonstrated with this analysis was consistent with expected outcome based on epidemiological data available. A brief description and instructions to aid interpretation of phylogenetic trees were included in the deployed standard operating procedure to enable local interpretation by clinicians. Deployed team members correctly interpreted both the phylogenetic tree to determine relatedness between the virus in specimens sequenced and single nucleotide polymorphism (SNP) analysis to determine variant type. This was in the absence of any direct available reach-back support during this phase of the study.
{kind=link}
{kind=link}
{kind=link}
Sequence relatedness was inferred using maximum likelihood method constructed using MEGAX analysis toolkit. The tree with the most significant log likelihood (−103157.09) is shown. The general-time-reversible model of evolution was used, and default tree branch settings used along with bootstrap analysis set to 100. Initial alignment of sequences was performed using MUSCLE.
Discussion
WGS of SARS-CoV-2 for epidemiological purposes, which has been in use since the outbreak began, has been demonstrably valuable and effective in the investigation of transmission chains not amenable to traditional methods. The ARTIC Network developed sequencing protocol used globally for the generation of genomic data for SARS-CoV-2, and has helped to identify the emergence and spread of new virus variants.25 The ability to genotype SARS-CoV-2 has become even more important following recognition that different variants of the virus demonstrate variable susceptibility to neutralisation with available medical countermeasures. For example, the Omicron variant possesses mutations in the receptor-binding domain of the viral spike glycoprotein which reduces susceptibility to neutralisation with the monoclonal antibodies casirivimab and imdevimab.26 Emergent subvariants of Omicron classified as BA.1 and BA.2 also display different susceptibility to the monoclonal antibody sotrovimab.27 Continuing emergence of new variants strengthens the requirement for genotyping capability to aid both direct patient management and to monitor the evolution of SARS-CoV-2.
This is particularly relevant to military populations and especially naval personnel who may be exposed to novel, emerging viral variants when visiting foreign ports after prolonged periods at sea. WGS data from this sentinel population would be valuable not only for charting virus evolution but also for identifying unusual clinical presentations that may be associated with new variants. Furthermore, as described earlier, transmission of SARS-CoV-2 can occur rapidly on ships at sea where many hundreds to thousands of personnel may be deployed, and implementation of effective infection control measures is difficult. If ongoing virus transmission occurs, which may be identified from new symptomatic individuals or asymptomatic cases following screening, despite the use of outbreak control measures, WGS has the potential to understand the routes of ongoing transmission through delineation of virus relatedness. This could facilitate the implementation of targeted measures for mitigation.
While use of target-specific WGS in remote environments has been demonstrated previously, the development of the Oxford Nanopore Technologies MinION was a milestone in widening access to next-generation sequencing technologies and enabling pathogen sequencing outside of traditional laboratory settings. The technology is sufficiently robust to allow deployment to rural and remote locations in low/middle-income countries and has been the cornerstone of sequencing in outbreak investigations and response, such as during the Ebola virus outbreak in West Africa in 2014 and in the subsequent outbreaks of Lassa fever virus. In both outbreaks, WGS identified transmission chains which traditional contact tracing approaches had not.28 29
The use of pathogen-specific WGS technology in a military medical setting is emerging30 and to our knowledge, its use aboard a military maritime capability has not previously been demonstrated. This proof-of-concept study has shown it is both functionally amenable and potentially useful even in situations where access to reach-back support and online hosted analysis tools is restricted.
While we have demonstrated feasibility of the technology, there are several important considerations about the practicalities of deployment, including ensuring access to the appropriate skillsets for analysing sequencing data and its integration into infection control strategies. We envisage this capability would be useful during a large deployment, such as maritime task group, which typically includes several ships, and has a significant population at risk of over a thousand personnel. Such deployments would usually contain an enhanced medical treatment facility which could include a military infection specialist with the skills and experience to initiate sequencing and interpret the data.
Furthermore, it is important to highlight the difference between pathogen-specific, targeted sequencing employed in this study and unbiased, culture-independent approaches which aim to identify pathogens that may be present at very low levels in clinical samples and require several technical challenges to be overcome. While we have used pathogen-specific genomics, nanopore sequencing technology can also be used for pathogen-agnostic metagenomic sequencing of specimens to identify novel and unexpected pathogens and biological threats. This study may help the development of future deployed metagenomics capability which could contribute to biological threat detection and control programmes in military settings.
Data availability statement
Data are available upon reasonable request. Data are available on reasonable request.
Ethics statements
Patient consent for publication
Ethics approval
Not applicable.
Acknowledgments
We acknowledge technical support from Dr Joshua Quick, University of Birmingham, UK, and logistical support from Captain Michael Brewer, Centre for Defence Pathology, Birmingham, UK.
References
Footnotes
Contributors MO'S conceived the study. AB, JR and CH undertook the experimental work. AB, JR and MO'S performed the data analysis. AB and MO'S wrote the first draft of the manuscript, and critical appraisal and revisions were made by all remaining authors. The final version of the manuscript was agreed by all authors. MO'S acts as the guarantor.
Funding Funding was provided by Research and Clinical Innovation, Defence Medical Directorate, Birmingham, UK (grant no. RCI/MR3WGS/01).
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.